
Épidémiologie et critères diagnostiques
La neurofibromatose de type 1 (NF1) est une maladie génétique de transmission autosomique dominante, liée à une mutation du gène NF1 codant la neurofibromine.
La génétique des neurofibromatoses est reprise dans un chapitre dédié. Sa prévalence est de 1/40001–4 et son incidence estimée à une naissance sur 30001,2,5–7.
Son expression phénotypique est variable même entre les membres d’une famille. Sa pénétrance est complète à l’âge de 8 ans : 97% des patients atteints de NF1 remplissent les critères diagnostiques à 8 ans8.
Le diagnostic repose sur l’association d’au moins 2 des 7 critères du NIH de 19889 révisés en 202110, et résumés ci-dessous. Ils diffèrent selon si le patient présente un de ses deux parents également atteints.
CRITÈRES DIAGNOSTIQUES DE LA NF1 – CONFÉRENCE DE CONSENSUS DU NIH DE 1988, RÉVISÉS EN 2021
A. En l’absence de parent atteint de NF1, le diagnostic de NF1 est posé chez un individu lorsqu’au moins deux critères sont présents parmi les suivants :
- Au moins six TCL de plus de 5 mm de diamètre chez un enfant pré-pubère et de plus de 15 mm chez un individu pubère.
- Des lentigines dans les plis axillaires et/ou inguinaux* et **.
- Au moins deux neurofibromes quel qu’en soit le type, ou un neurofibrome plexiforme.
- Un gliome des voies optiques.
- Au moins deux nodules de Lisch identifiés à l’examen à la lampe à fente, ou au moins deux Anomalies Choroïdiennes (CA), définies comme des taches hyper-réflectives mises en évidence en Tomographie par Cohérence Optique (OCT), sur les clichés en proche infra-rouge (NIR).
- Une lésion osseuse identifiée parmi les suivantes : dysplasie du sphénoïde***, courbure antérolatérale du tibia ou pseudarthrose d’un os long.
- Un variant pathogène hétérozygote du gène NF1, avec une fraction allélique de 50% dans un tissu apparemment normal tel que les globules blancs.
B. Chez un enfant qui présente un parent répondant aux critères diagnostiques de NF1 spécifiés en A, la présence d’au moins un critère de A permet de poser le diagnostic de NF1.
**Au moins un des deux types de lésions pigmentées (TCL ou lentigines) doit être de topographie bilatérale.
***La dysplasie d’une aile sphénoïdale ne constitue pas un critère indépendant en cas de neurofibrome plexiforme orbitaire homolatéral.
-
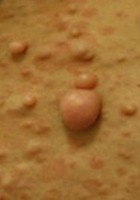
- Neurofibromes cutanés (adulte)
-
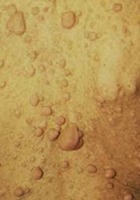
- Neurofibromes cutanés (adulte)
-
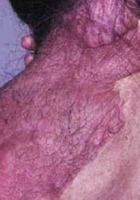
- Neurofibrome plexiforme cervical postérieur (adulte)
-
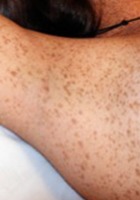
- Lentigines axillaires (adulte)
-
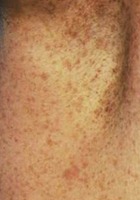
- Lentigines axillaires (adulte)
-
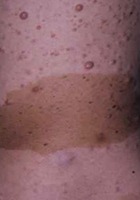
- Tache café au lait associée à des neurofibromes cutanés (adulte)
-
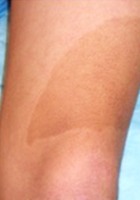
- Large tache café au lait (adulte)
-
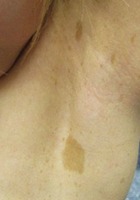
- Taches café au lait associées à des lentigines axillaires bilatérales (enfant)
-
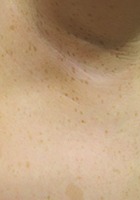
- Taches café au lait associées à des lentigines axillaires bilatérales (enfant)
-
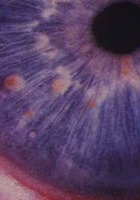
- Nodules de Lisch (examen à la lampe à fente)
Complications
Les complications de la maladie sont nombreuses, bénignes ou malignes, somatiques et psychologiques, et apparaissent tout au long de la vie11. Cependant, une des particularités de la NF1 reste sa variabilité d’expression d’un patient à un autre et même au sein d’une famille (où l’anomalie génétique est pourtant identique). Ceci rend impossible de prédire l’évolution clinique d’un enfant atteint.
Les neurofibromes peuvent être de différents types : dermiques (plus de 95% des patients)12,13, nodulaires périphériques (au moins 20% des patients)12, nodulaires, ou plexiformes (20 % des patients)14. Le risque principal, inhérent aux neurofibromes nodulaires et plexiformes, est la Transformation en Tumeur Maligne des Gaines Nerveuses périphériques (TMGN, anciennement tumeur maligne des gaines nerveuses). Le risque cumulé de développer une TMGN au cours de la vie d’un patient a été estimé à 8-16%. Il survient majoritairement entre 20 et 35 ans15–19.
Ce risque est majoré chez les patients présentant un phénotype de NF1 à risque défini par la présence de neurofibromes sous-cutanés : en cas de neurofibromes sous-cutanés, le risque de développer un neurofibrome plexiforme interne ou une TMGN est multiplié par 3, et en cas de neurofibrome plexiforme, le risque de développer une TMGN est multiplié par 2020. La tumeur peut être agressive17 , avec une espérance de vie diminuée16.
Les imageries métaboliques comme par PET-IRM ou le PET-scanner peuvent diagnostiquer le neurofibrome au stade dysplasique et ainsi améliorer le pronostic par une chirurgie précoce. Les neurofibromes peuvent aussi avoir un retentissement esthétique et fonctionnel non négligeable.
Le gliome des voies optiques21–25 est une tumeur bénigne, au faible potentiel de malignité, présente chez 15 à 20% des enfants atteints de NF121, 26–30. En revanche, son extension locale peut entraîner une exophtalmie, une baisse de l’acuité visuelle ou une compression du chiasma optique avec d’éventuels retentissements sur l’axe hypothalamo-hypophysaire gonadotrope et une puberté précoce30, 31.
Les atteintes orthopédiques sont classiques : dysplasies congénitales des os longs (7,2% des patients)32, dysplasie sphénoïdale (1 à 7% des patients)33 et scoliose (10 à 28% des patients)34.
Les dysplasies congénitales des os longs entrainent des déformations, et un sur-risque de fracture avec constitution de pseudarthrose chez 2 à 3.6% des patients32, 35.
La dysplasie sphénoïdale peut être secondaire à un neurofibrome plexiforme36, peut entrainer une exophtalmie pulsatile sans baisse de l’acuité visuelle et une hernie du lobe temporal dans le globe oculaire en cas d’absence totale d’os sphénoïdal37.
La scoliose, manifestation orthopédique la plus fréquente, est souvent associée à une dysplasie vertébrale38.
Les patients atteints de NF1 semblent présenter un risque accru d’ostéopathie fragilisante avec une ostéopénie chez 48% des patients ou une ostéoporose chez 25%39, 40.
De manière précoce, un retard de croissance est présent chez un tiers des enfants41, une puberté précoce peut également survenir33, 42. La puberté précoce se définit par l’apparition de critères sexuels avant l’âge de 7-8 ans chez la fille et 9-10 ans chez le garçon.
La complication neurologique centrale la plus sévère est l’épilepsie et ses répercussions développementales. Elle touche 8% des patients56, 57, est résistante au traitement dans 30% des cas58. Dans ces derniers cas, elle peut s’associer à un retard mental sévère.
Les troubles cognitifs modérés, en dehors de toute épilepsie, constituent la complication neurologique la plus fréquente59, avec un quotient intellectuel dans la partie moyenne basse, des troubles du comportement et des difficultés d’apprentissages spécifiques (troubles visuo-spatiaux et visuo-moteurs, troubles du langage, troubles de la motricité fine et brute, troubles des fonctions exécutives)5, 59–64. D’autres pathologies ont été associées à la NF1 : trouble de déficit de l’attention/hyperactivité, troubles autistiques, troubles psychosociaux62, 64.
Enfin, les patients atteints de NF1 ont un risque accru de néoplasie65.
Concernant le risque de cancer du sein, particulièrement étudié dans la population de patients atteints de NF1, il survient chez des femmes de moins de 50 ans66–70 avec un taux de mortalité plus important71. Ces caractéristiques justifient un dépistage systématique annuel par l’association d’une mammographie-échographie72 et d’une IRM mammaire, à partir de l’âge de 30 ans chez les femmes atteintes de NF1.
Les TMGN représentent 60% des cancers dans cette population mais les patients peuvent également développer d’autres tumeurs du spectre neurologique : les tumeurs cérébrales, dont les gliomes des voies optiques, mais aussi les gliomes du tronc cérébral73, les tumeurs gastro-intestinales neuroendocrines qui surviennent souvent à un âge plus jeune que dans la population générale, sont souvent multiples et se situent régulièrement dans l’intestin grêle74, 75.
Les répercussions psychologiques chroniques de la NF1 ont fait l’objet de deux études évaluant les troubles anxio-dépressifs chez l’adulte76, et chez l’enfant77 : 19% des adultes rapportent des symptômes dépressifs, 15% des troubles anxieux76. Chez l’adulte, les associations entre la sévérité des symptômes anxio-dépressifs et la sévérité de la maladie (calculée par une version modifiée du score de Riccardi78) ou la visibilité des lésions cutanées (calculée selon le score de Ablon79) sont significatives (p<0.001)76.
Les perturbations du sommeil chez les patients atteints de NF1 ont également été évaluées dans une étude chez l’adulte80 et une chez l’enfant64. Les moyennes des scores ESS (Epworth Sleepiness Scale)81 et PSQI (Pittsburgh Sleep Quality Index)82 évaluant les perturbations du sommeil sont anormalement élevées chez ces patients80, sans corrélation entre les deux scores.
Chez les enfants atteints de NF1, les problèmes de conduite (p ≤ 0.05), l’hyperactivité (p ≤ 0.01), la fragilité émotionnelle (p ≤ 0.01) et le score total de difficultés (p ≤ 0.01) sont significativement augmentés et corrélés aux perturbations du sommeil64. Ces retentissements psychologiques et sur le sommeil, associés aux douleurs chroniques, contribuent largement à l’altération de la qualité de vie des patients atteints de NF1 83–85. Une adaptation des scores de qualité de vie (Skindex-16) et du fardeau sont en cours pour la NF1 : cNF-SKindex86 et Burden of NF1 respectivement.
La prise en charge au long cours de ces patients doit intégrer cette dimension.
Le risque de mortalité est accru chez les patients atteints de NF1. En comparaison à la population générale, leur espérance de vie est raccourcie de 10 à 15 ans15, 66, 67, 71, 87–89. En France, la dernière étude de la mortalité au cours de la NF1 date de 201189, et concernait les données de 1985 patients entre 1980 et 2005.
La mortalité toutes causes confondues était significativement plus élevée dans la cohorte de patients atteints de NF1 avec un SMR (Standardized Mortality Ratio) de 2.02 (1.6-2.6) (p<0.0001). L’étude de la cause des décès, disponible pour 58 patients (86.6% des patients décédés), retrouvait les TMGN comme principale cause.
Principal diagnostic différentiel
Le principal diagnostic différentiel de la neurofibromatose de type 1 (NF1) est le syndrome de Legius. Tout comme les critères diagnostiques de NF1, ceux du syndrome de Legius se déclinent en deux situations (A et B) selon l’absence (A) ou la présence (B) d’un parent atteint.
Ainsi, le diagnostic de syndrome de Legius est posé dans les conditions suivantes :
A. En l’absence de parent atteint de syndrome de Legius, le diagnostic est posé chez un individu lorsque les critères suivants sont réunis :
- Au moins six TCL distribuées de manière bilatérale, en l’absence d’autre critère diagnostique de NF1 hormis la présence de lentigines axillaires ou inguinales*.
- Un variant pathogène hétérozygote du gène SPRED1, avec une fraction allélique de 50% dans un tissu apparemment normal tel que les globules blancs.
Prise en charge
Dans ses formes les plus symptomatiques, la NF1 nécessite un suivi pluridisciplinaire et pluri professionnel, au long cours avec une recherche spécifique des différentes complications citées préalablement et un étayage adapté parfois dès le plus jeune âge (avec un accompagnement des apprentissages par exemple).
Cette prise en charge s’organise au quotidien à proximité des patients avec recours quand nécessaire au centre de référence des neurofibromatoses.
Le PNDS a été mis à jour en 2021, vous le trouverez sur le site internet de la Haute Autorité de Santé ou sur la page des documents pratiques pour les professionnels de santé.
S’il n’existe pas de traitement de la mutation génétique responsable de la NF1, il existe en revanche de multiples traitements des différentes manifestations et complications de la maladie.
Ces complications ne sont le plus souvent pas spécifiques de la NF1 et leurs prises en charge ne diffèrent donc pas des mêmes situations rencontrées chez des patients non atteints de NF1. Plusieurs situations sont néanmoins à préciser.
La douleur est un symptôme très fréquent au cours de la NF1 et peut accompagner voire révéler de nombreuses complications. La composante neuropathique de la douleur est prépondérante lorsqu’elle est secondaire à des neurofibromes internes compressifs et/ou transformés. L’utilisation, en plus d’antalgiques classiques, de traitements actifs sur les douleurs neuropathiques est donc essentielle au cours de la NF1. Il peut s’agir de médicaments de la famille des antiépileptiques et/ou des antidépresseurs.
La gestion des douleurs peut être améliorée par l’apprentissage de techniques non médicamenteuses, notamment lorsque celles-ci deviennent chroniques et rebelles aux traitements « classiques ». Ainsi, l’aide d’équipes spécialisées dans l’étude et le traitement des douleurs est parfois nécessaire. Enfin, il ne faut pas négliger le retentissement émotionnel négatif de ces douleurs, ce qui peut conduire à la mise en place d’un soutien psychologique adapté.
Pour les patients adultes, de nombreuses structures hospitalières proposent une prise en charge spécialisée de la douleur. Les centres de référence et compétences travaillent le plus souvent avec une équipe de prise en charge de la douleur, du même hôpital. Pour les patients franciliens, souffrant d’une douleur chronique, ils peuvent s’adresser à une structure spécialisée au sein de l’AP-HP.
L’évaluation et la prise en charge de la douleur de l’enfant sont complexes et font appel le plus souvent à des équipes multidisciplinaires et pluri-professionnelles. Des sites internet permettent d’aider les parents des enfants mais également les professionnels de santé dans l’évaluation et la prise en charge de la douleur comme Pediadol ou Dolomio, groupe d’experts de la douleur de l’enfant et de l’adolescent.
Le recours à une équipe spécialisée peut parfois être nécessaire, ainsi, les centres de référence et de compétences pour la prise en charge des enfants atteints de NF1 travaillent souvent en étroite collaboration avec des équipes spécialisées dans le traitement de la douleur.
Les Tumeurs Malignes des Gaines Nerveuses (TMGN) sont des complications graves dont le pronostic dépend de la possibilité d’en effectuer une exérèse chirurgicale large. Après le traitement chirurgical quand il est possible, un traitement adjuvant par radiothérapie est le plus souvent proposé. En cas de maladie localement avancée et inopérable, ou au stade métastatique, des poly-chimiothérapies sont proposées.
Les neurofibromes cutanés ne sont pas à risque de transformation en TMGN mais ils peuvent être responsables d’une gêne esthétique majeure. Les seuls traitements actuellement disponibles sont des traitements interventionnels : laser CO2, exérèse chirurgicale classique, électro-dissection. Les résultats esthétiques dépendent de la qualité de la cicatrisation et du nombre de lésions à traiter.
Les inhibiteurs de MEK ont émergé récemment dans le cadre de la NF1, notamment le tramétinib et le sélumétinib. Plusieurs études ont montré leur efficacité comme traitement des neurofibromes internes symptomatiques et inopérables90–93. Récemment, les élumétinib a obtenu une Autorisation de Mise sur le Marché (AMM), à l’échelle européenne, pour l’indication suivante : les neurofibromes plexiformes inopérables de l’enfant.
Cette AMM remplace les Autorisations Temporaires d’Utilisation (ATU) initialement requises. Ces traitements peuvent être prescrits dans des centres spécialisés après discussion collégiale. Les données d’utilisation en vie réelle et le développement de nouvelles molécules et formulations pourraient permettre d’étendre leurs champs d’utilisation dans le futur.
Bibliographie
- Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005;141(1):71-74. doi:10.1001/archderm.141.1.71.
- Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152A(2):327-332. doi:10.1002/ajmg.a.33139.
- Huson SM, Compston DA, Clark P, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989;26(11):704-711. doi:10.1136/jmg.26.11.704.
- Kallionpää RA, Uusitalo E, Leppävirta J, Pöyhönen M, Peltonen S, Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet Med Off J Am Coll Med Genet. 2018;20(9):1082-1086. doi:10.1038/gim.2017.215.
- Huson SM, Compston DA, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. II. Guidelines for genetic counselling. J Med Genet. 1989;26(11):712-721. doi:10.1136/jmg.26.11.712.
- Poyhonen M, Kytölä S, Leisti J. Epidemiology of neurofibromatosis type 1 (NF1) in northern Finland. J Med Genet. 2000;37(8):632-636. doi:10.1136/jmg.37.8.632.
- Uusitalo E, Leppävirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015;135(3):904-906. doi:10.1038/jid.2014.465.
- DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105(3 Pt 1):608-614. doi:10.1542/peds.105.3.608.
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45(5):575-578.
- Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Do G. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. Published online May 19, 2021. doi:10.1038/s41436-021-01170-5
- Bergqvist C, Servy A, Valeyrie-Allanore L, et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis. 2020;15(1):37. doi:10.1186/s13023-020-1310-3.
- Duong TA, Bastuji-Garin S, Valeyrie-Allanore L, Sbidian E, Ferkal S, Wolkenstein P. Evolving pattern with age of cutaneous signs in neurofibromatosis type 1: a cross-sectional study of 728 patients. Dermatol Basel Switz. 2011;222(3):269-273. doi:10.1159/000327379.
- Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: Current clinical and pathologic issues. Neurology. 2018;91(2 Suppl 1):S5-S13. doi:10.1212/WNL.0000000000005792.
- Darrigo LG, Geller M, Bonalumi Filho A, Azulay DR. Prevalence of plexiform neurofibroma in children and adolescents with type I neurofibromatosis. J Pediatr (Rio J). 2007;83(6):571-573. doi:10.2223/JPED.1718.
- Uusitalo E, Rantanen M, Kallionpää RA, et al. Distinctive Cancer Associations in Patients With Neurofibromatosis Type 1. J Clin Oncol Off J Am Soc Clin Oncol. 2016;34(17):1978-1986. doi:10.1200/JCO.2015.65.3576.
- Evans DGR, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311-314. doi:10.1136/jmg.39.5.311.
- Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62(5):1573-1577.
- Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006-2021. doi:10.1002/1097-0142(19860515)57:10<2006::aid-cncr2820571022>3.0.co;2-6.
- Evans DGR, Huson SM, Birch JM. Malignant peripheral nerve sheath tumours in inherited disease. Clin Sarcoma Res. 2012;2(1):17. doi:10.1186/2045-3329-2-17.
- Tucker T, Wolkenstein P, Revuz J, Zeller J, Friedman JM. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology. 2005;65(2):205-211. doi:10.1212/01.wnl.0000168830.79997.13.
- Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63-66. doi:10.1016/s0022-3476(94)70122-9.
- Slamovits TL. Von Recklinghausen neurofibromatosis: II. Incidence of optic gliomata. Ophthalmology. 1985;92(5):714-715.
- Kornreich L, Blaser S, Schwarz M, et al. Optic pathway glioma: correlation of imaging findings with the presence of neurofibromatosis. AJNR Am J Neuroradiol. 2001;22(10):1963-1969.
- Deliganis AV, Geyer JR, Berger MS. Prognostic significance of type 1 neurofibromatosis (von Recklinghausen Disease) in childhood optic glioma. Neurosurgery. 1996;38(6):1114-1118; discussion 1118-1119. doi:10.1097/00006123-199606000-00010.
- Listernick R, Darling C, Greenwald M, Strauss L, Charrow J. Optic pathway tumors in children: the effect of neurofibromatosis type 1 on clinical manifestations and natural history. J Pediatr. 1995;127(5):718-722. doi:10.1016/s0022-3476(95)70159-1.
- Listernick R, Charrow J, Greenwald MJ, Esterly NB. Optic gliomas in children with neurofibromatosis type 1. J Pediatr. 1989;114(5):788-792. doi:10.1016/s0022-3476(89)80137-4.
- Lewis RA, Gerson LP, Axelson KA, Riccardi VM, Whitford RP. von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology. 1984;91(8):929-935. doi:10.1016/s0161-6420(84)34217-8.
- Lund AM, Skovby F. Optic gliomas in children with neurofibromatosis type 1. Eur J Pediatr. 1991;150(12):835-838. doi:10.1007/BF01955002.
- Prada CE, Hufnagel RB, Hummel TR, et al. The Use of Magnetic Resonance Imaging Screening for Optic Pathway Gliomas in Children with Neurofibromatosis Type 1. J Pediatr. 2015;167(4):851-856.e1. doi:10.1016/j.jpeds.2015.07.001.
- Blazo MA, Lewis RA, Chintagumpala MM, Frazier M, McCluggage C, Plon SE. Outcomes of systematic screening for optic pathway tumors in children with Neurofibromatosis Type 1. Am J Med Genet A. 2004;127A(3):224-229. doi:10.1002/ajmg.a.20650.
- Laue L, Comite F, Hench K, Loriaux DL, Cutler GB, Pescovitz OH. Precocious puberty associated with neurofibromatosis and optic gliomas. Treatment with luteinizing hormone releasing hormone analogue. Am J Dis Child 1960. 1985;139(11):1097-1100. doi:10.1001/archpedi.1985.02140130035025.
- Boulanger JM, Larbrisseau A. Neurofibromatosis type 1 in a pediatric population: Ste-Justine’s experience. Can J Neurol Sci J Can Sci Neurol. 2005;32(2):225-231. doi:10.1017/s0317167100004017.
- Pinson S, Créange A, Barbarot S, et al. [Recommendations for the treatment of neurofibromatosis type 1]. J Fr Ophtalmol. 2002;25(4):423-433.
- Akbarnia BA, Gabriel KR, Beckman E, Chalk D. Prevalence of scoliosis in neurofibromatosis. Spine. 1992;17(8 Suppl):S244-248. doi:10.1097/00007632-199208001-00005.
- Ferner RE. Neurofibromatosis 1. Eur J Hum Genet EJHG. 2007;15(2):131-138. doi:10.1038/sj.ejhg.5201676.
- Arrington DK, Danehy AR, Peleggi A, Proctor MR, Irons MB, Ullrich NJ. Calvarial defects and skeletal dysplasia in patients with neurofibromatosis Type 1. J Neurosurg Pediatr. 2013;11(4):410-416. doi:10.3171/2013.1.PEDS12409.
- Ferner RE. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol. 2007;6(4):340-351. doi:10.1016/S1474-4422(07)70075-3.
- Ramachandran M, Tsirikos AI, Lee J, Saifuddin A. Whole-spine magnetic resonance imaging in patients with neurofibromatosis type 1 and spinal deformity. J Spinal Disord Tech. 2004;17(6):483-491. doi:10.1097/01.bsd.0000133466.97241.50.
- Brunetti-Pierri N, Doty SB, Hicks J, et al. Generalized metabolic bone disease in Neurofibromatosis type I. Mol Genet Metab. 2008;94(1):105-111. doi:10.1016/j.ymgme.2007.12.004.
- Lammert M, Kappler M, Mautner V-F, et al. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA. 2005;16(9):1161-1166. doi:10.1007/s00198-005-1940-2.
- Vassilopoulou-Sellin R, Woods D, Quintos MT, Needle M, Klein MJ. Short stature in children and adults with neurofibromatosis. Pediatr Nurs. 1995;21(2):149-153.
- Virdis R, Sigorini M, Laiolo A, et al. Neurofibromatosis type 1 and precocious puberty. J Pediatr Endocrinol Metab JPEM. 2000;13 Suppl 1:841-844. doi:10.1515/jpem.2000.13.s1.841.
- Lama G, Graziano L, Calabrese E, et al. Blood pressure and cardiovascular involvement in children with neurofibromatosis type1. Pediatr Nephrol Berl Ger. 2004;19(4):413-418. doi:10.1007/s00467-003-1397-5.
- Fossali E, Signorini E, Intermite RC, et al. Renovascular disease and hypertension in children with neurofibromatosis. Pediatr Nephrol Berl Ger. 2000;14(8-9):806-810. doi:10.1007/s004679900260.
- Virdis R, Balestrazzi P, Zampolli M, Donadio A, Street M, Lorenzetti E. Hypertension in children with neurofibromatosis. J Hum Hypertens. 1994;8(5):395-397.
- Tedesco MA, Di Salvo G, Ratti G, et al. Arterial distensibility and ambulatory blood pressure monitoring in young patients with neurofibromatosis type 1. Am J Hypertens. 2001;14(6 Pt 1):559-566. doi:10.1016/s0895-7061(00)01303-0.
- Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med Off J Am Coll Med Genet. 2002;4(3):105-111. doi:10.1097/00125817-200205000-00002.
- Neiman HL, Mena E, Holt JF, Stern AM, Perry BL. Neurofibromatosis and congenital heart disease. Am J Roentgenol Radium Ther Nucl Med. 1974;122(1):146-149. doi:10.2214/ajr.122.1.146.
- Holt JF. 1977 Edward B. D. Neuhauser lecture: neurofibromatosis in children. AJR Am J Roentgenol. 1978;130(4):615-639. doi:10.2214/ajr.130.4.615.
- Carey JC, Laub JM, Hall BD. Penetrance and variability in neurofibromatosis: a genetic study of 60 families. Birth Defects Orig Artic Ser. 1979;15(5B):271-281.
- Schorry EK, Stowens DW, Crawford AH, Stowens PA, Schwartz WR, Dignan PS. Summary of patient data from a multidisciplinary neurofibromatosis clinic. Neurofibromatosis. 1989;2(2):129-134.
- Colley A, Donnai D, Evans DG. Neurofibromatosis/Noonan phenotype: a variable feature of type 1 neurofibromatosis. Clin Genet. 1996;49(2):59-64. doi:10.1111/j.1399-0004.1996.tb04328.x.
- Tonsgard JH, Yelavarthi KK, Cushner S, Short MP, Lindgren V. Do NF1 gene deletions result in a characteristic phenotype? Am J Med Genet. 1997;73(1):80-86. doi:10.1002/(sici)1096-8628(19971128)73:1<80::aid-ajmg16>3.0.co;2-n.
- Ademiluyi SA, Sowemimo GO, Oyeneyin JO. Surgical experience in the management of multiple neurofibromatosis in Nigerians. West Afr J Med. 1989;8(1):59-65.
- Rasko JE, North KN, Favaloro EJ, Grispo L, Berndt MC. Attenuated platelet sensitivity to collagen in patients with neurofibromatosis type 1. Br J Haematol. 1995;89(3):582-588. doi:10.1111/j.1365-2141.1995.tb08367.x.
- Korf BR, Carrazana E, Holmes GL. Patterns of seizures observed in association with neurofibromatosis 1. Epilepsia. 1993;34(4):616-620. doi:10.1111/j.1528-1157.1993.tb00437.x.
- Kulkantrakorn K, Geller TJ. Seizures in neurofibromatosis 1. Pediatr Neurol. 1998;19(5):347-350. doi:10.1016/s0887-8994(98)00075-7.
- Holmes MD. The causes of epilepsy: common and uncommon causes in adults and children. Arch Neurol. 2012;69(9):1212-1213. doi:10.1001/archneurol.2012.1266.
- North KN, Riccardi V, Samango-Sprouse C, et al. Cognitive function and academic performance in neurofibromatosis. 1: consensus statement from the NF1 Cognitive Disorders Task Force. Neurology. 1997;48(4):1121-1127. doi:10.1212/wnl.48.4.1121.
- Descheemaeker M-J, Plasschaert E, Frijns J-P, Legius E. Neuropsychological profile in adults with neurofibromatosis type 1 compared to a control group. J Intellect Disabil Res JIDR. 2013;57(9):874-886. doi:10.1111/j.1365-2788.2012.01648.x.
- Ferner RE, Hughes RA, Weinman J. Intellectual impairment in neurofibromatosis 1. J Neurol Sci. 1996;138(1-2):125-133. doi:10.1016/0022-510x(96)00022-6.
- Acosta MT, Gioia GA, Silva AJ. Neurofibromatosis type 1: new insights into neurocognitive issues. Curr Neurol Neurosci Rep. 2006;6(2):136-143. doi:10.1007/s11910-996-0036-5.
- Hyman SL, Arthur Shores E, North KN. Learning disabilities in children with neurofibromatosis type 1: subtypes, cognitive profile, and attention-deficit-hyperactivity disorder. Dev Med Child Neurol. 2006;48(12):973-977. doi:10.1017/S0012162206002131.
- Johnson H, Wiggs L, Stores G, Huson SM. Psychological disturbance and sleep disorders in children with neurofibromatosis type 1. Dev Med Child Neurol. 2005;47(4):237-242. doi:10.1017/s0012162205000460.
- Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: population-based record-linkage study. Br J Cancer. 2013;108(1):193-198. doi:10.1038/bjc.2012.535.
- Madanikia SA, Bergner A, Ye X, Blakeley JO. Increased risk of breast cancer in women with NF1. Am J Med Genet A. 2012;158A(12):3056-3060. doi:10.1002/ajmg.a.35550.
- Walker L, Thompson D, Easton D, et al. A prospective study of neurofibromatosis type 1 cancer incidence in the UK. Br J Cancer. 2006;95(2):233-238. doi:10.1038/sj.bjc.6603227.
- Wang X, Levin AM, Smolinski SE, Vigneau FD, Levin NK, Tainsky MA. Breast cancer and other neoplasms in women with neurofibromatosis type 1: a retrospective review of cases in the Detroit metropolitan area. Am J Med Genet A. 2012;158A(12):3061-3064. doi:10.1002/ajmg.a.35560.
- Sharif S, Moran A, Huson SM, et al. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet. 2007;44(8):481-484. doi:10.1136/jmg.2007.049346.
- Uusitalo E, Kallionpää RA, Kurki S, et al. Breast cancer in neurofibromatosis type 1: overrepresentation of unfavourable prognostic factors. Br J Cancer. 2017;116(2):211-217. doi:10.1038/bjc.2016.403.
- Evans DGR, O’Hara C, Wilding A, et al. Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet EJHG. 2011;19(11):1187-1191. doi:10.1038/ejhg.2011.113.
- Daly MB, Pilarski R, Berry M, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J Natl Compr Cancer Netw JNCCN. 2017;15(1):9-20. doi:10.6004/jnccn.2017.0003.
- Guillamo J-S, Créange A, Kalifa C, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain J Neurol. 2003;126(Pt 1):152-160. doi:10.1093/brain/awg016.
- Salvi PF, Lorenzon L, Caterino S, Antolino L, Antonelli MS, Balducci G. Gastrointestinal stromal tumors associated with neurofibromatosis 1: a single centre experience and systematic review of the literature including 252 cases. Int J Surg Oncol. 2013;2013:398570. doi:10.1155/2013/398570.
- Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30(1):90-96. doi:10.1097/01.pas.0000176433.81079.bd.
- Doser K, Andersen EW, Kenborg L, et al. Clinical characteristics and quality of life, depression, and anxiety in adults with neurofibromatosis type 1: A nationwide study. Am J Med Genet A. 2020;182(7):1704-1715. doi:10.1002/ajmg.a.61627.
- Pasini A, Lo-Castro A, Di Carlo L, et al. Detecting anxiety symptoms in children and youths with neurofibromatosis type I. Am J Med Genet Part B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet. 2012;159B(7):869-873. doi:10.1002/ajmg.b.32095.
- Barton B, North K. The self-concept of children and adolescents with neurofibromatosis type 1. Child Care Health Dev. 2007;33(4):401-408. doi:10.1111/j.1365-2214.2006.00717.x.
- Ablon J. Gender response to neurofibromatosis 1. Soc Sci Med 1982. 1996;42(1):99-109. doi:10.1016/0277-9536(95)00076-3.
- Leschziner GD, Golding JF, Ferner RE. Sleep disturbance as part of the neurofibromatosis type 1 phenotype in adults. Am J Med Genet A. 2013;161A(6):1319-1322. doi:10.1002/ajmg.a.35915.
- Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14(6):540-545. doi:10.1093/sleep/14.6.540.
- Buysse DJ, Reynolds CF, Monk TH, Berman SR, Kupfer DJ. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res. 1989;28(2):193-213. doi:10.1016/0165-1781(89)90047-4.
- Vranceanu A-M, Merker VL, Park E, Plotkin SR. Quality of life among adult patients with neurofibromatosis 1, neurofibromatosis 2 and schwannomatosis: a systematic review of the literature. J Neurooncol. 2013;114(3):257-262. doi:10.1007/s11060-013-1195-2.
- Vranceanu A-M, Merker VL, Park ER, Plotkin SR. Quality of life among children and adolescents with neurofibromatosis 1: a systematic review of the literature. J Neurooncol. 2015;122(2):219-228. doi:10.1007/s11060-015-1725-1.
- Garwood MM, Bernacki JM, Fine KM, Hainsworth KR, Davies WH, Klein-Tasman BP. Physical, cognitive, and psychosocial predictors of functional disability and health-related quality of life in adolescents with neurofibromatosis-1. Pain Res Treat. 2012;2012:975364. doi:10.1155/2012/975364.
- Fertitta L, Bergqvist C, Armand ML, et al. Quality of life in neurofibromatosis 1: development and validation of a tool dedicated to cutaneous neurofibromas in adults. J EurAcadDermatolVenereol 2022. doi:10.1111/jdv.18140.
- Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68(5):1110-1118. doi:10.1086/320121.
- Zöller M, Rembeck B, Akesson HO, Angervall L. Life expectancy, mortality and prognostic factors in neurofibromatosis type 1. A twelve-year follow-up of an epidemiological study in Göteborg, Sweden. Acta Derm Venereol. 1995;75(2):136-140. doi:10.2340/0001555575136140.
- Duong TA, Sbidian E, Valeyrie-Allanore L, et al. Mortality associated with neurofibromatosis 1: a cohort study of 1895 patients in 1980-2006 in France. Orphanet J Rare Dis. 2011;6:18. doi:10.1186/1750-1172-6-18.
- Dombi E, Baldwin A, Marcus LJ, et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. https://doi.org/10.1056/NEJMoa1605943. doi:10.1056/NEJMoa1605943.
- Weiss BD, Wolters PL, Plotkin SR, et al. NF106: A Neurofibromatosis Clinical Trials Consortium Phase II Trial of the MEK Inhibitor. Mirdametinib (PD-0325901) in Adolescents and Adults With NF1-Related Plexiform Neurofibromas. J Clin Oncol Off J Am Soc Clin Oncol. 2021;39(7):797-806. doi:10.1200/JCO.20.02220.
- Gross AM, Wolters PL, Dombi E, et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Engl J Med. 2020;382(15):1430-1442. doi:10.1056/NEJMoa1912735.
- Fisher MJ, Shih C-S, Rhodes SD, et al. Cabozantinib for neurofibromatosis type 1-related plexiform neurofibromas: a phase 2 trial. Nat Med. 2021;27(1):165-173. doi:10.1038/s41591-020-01193-6.